by Amélie Godard Palluet & Michał Żółtowski & Rafael Jara Toro & Sándor Demes & Cheikh Bop & François Lique, on
Presentation of the symposium
The Gas-Kinetics symposium was held in Rennes at Le Couvent des Jacobins, from the 28th of August, up to the 1st of September 2022.
It provided an open forum for the presentation and discussion of the latest research on a wide range of topics related to gas-phase chemical kinetics. This has been expanded over recent years to cover all aspects of atmospheric and combustion chemistry, as well as astrochemistry and planetary atmospheres, with heterogeneous systems involving micro- and nanoparticles getting an increasing role.
A. Godard Palluet presented her work in an oral communication.
R. Jara Toro, S. Demes and M. Żółtowski presented their work in a poster.
C. Bop and F. Lique also attended the symposium.
All of the present members of the team were also part of the organizing committee.
Fine and hyperfine excitation of CCS isotopologues induced by collisions with He - A. Godard Palluet and F. Lique

The physical conditions in space are derived from the spectra captured by telescopes. The spectral analysis requires to know the population of molecular energy levels. In astrophysical media, where the local thermodynamic equilibrium conditions are rarely fulfilled, the competition between radiative and collisional processes must to be taken into account. Rate coefficients, which characterize collisional processes, are hence used to derive the abundances of the chemical species. They are preferentially obtained from scattering calculations performed on a potential energy surface (PES) which describe the interaction between the two colliders.
The CCS(3Σ-) radical and its isotopologues have been detected in several dark molecular clouds, including TMC-1 and in the circumstellar enveloppe IRC+10216 [1,2]. The CCS radical presents large fine structure splitting, which makes the estimation of the rate coefficients incorrect and the line modeling difficult [1]. Therefore, the CCS rate coefficients must be accurately determined.
The determination of CCS abundances derived from these rate coefficients could provide rich informations. The abundance ratio between CCS and NH3 could characterize evolution stage of molecular clouds [3].
The PES used for the close coupling scattering calculations will be presented. It was obtained using the gold standard Coupled-Cluster method and an aVQZ basis set with additional mid-bond functions. The first accurate fine state-to-state rate coefficients for the 5 - 50 K range of temperature for the 12C12C32S isotopologue will be presented. These rates were also computed for the 13C12C32S, 12C13C32S and 12C12C34S isotopologues, and the isotopic effect will be discussed. The hyperfine structure of 13C-based isotopologues was taken into account in the calculations to provide rate coefficients for the 5 - 20 K temperature range. These hyperfine transitions were recently observed and these rate coefficients could help to understand the chemical processes forming the CCS radical [4].
[1] S. Saito, K. Kawaguchi, S. Yamamoto, M. Ohishi, H. Suzuki, N. Kaifu, ApJ., 317 L115-L119 (1987).
[2] H. Suzuki, S. Yamamoto, M. Ohishi, N. Kaifu, S. Ishikawa, Y. Hirahara, S. Takano, ApJ., 392 551-570 (1992).
[3] J. Cernicharo, M. Guélin, H. Hein, C. Kahane, A&A, 181 L9-L12 (1987).
[4] M. Ikeda, Y. Sekimoto, S. Yamamoto, J. Mol. Spectrosc. 185 21-25 (1997).
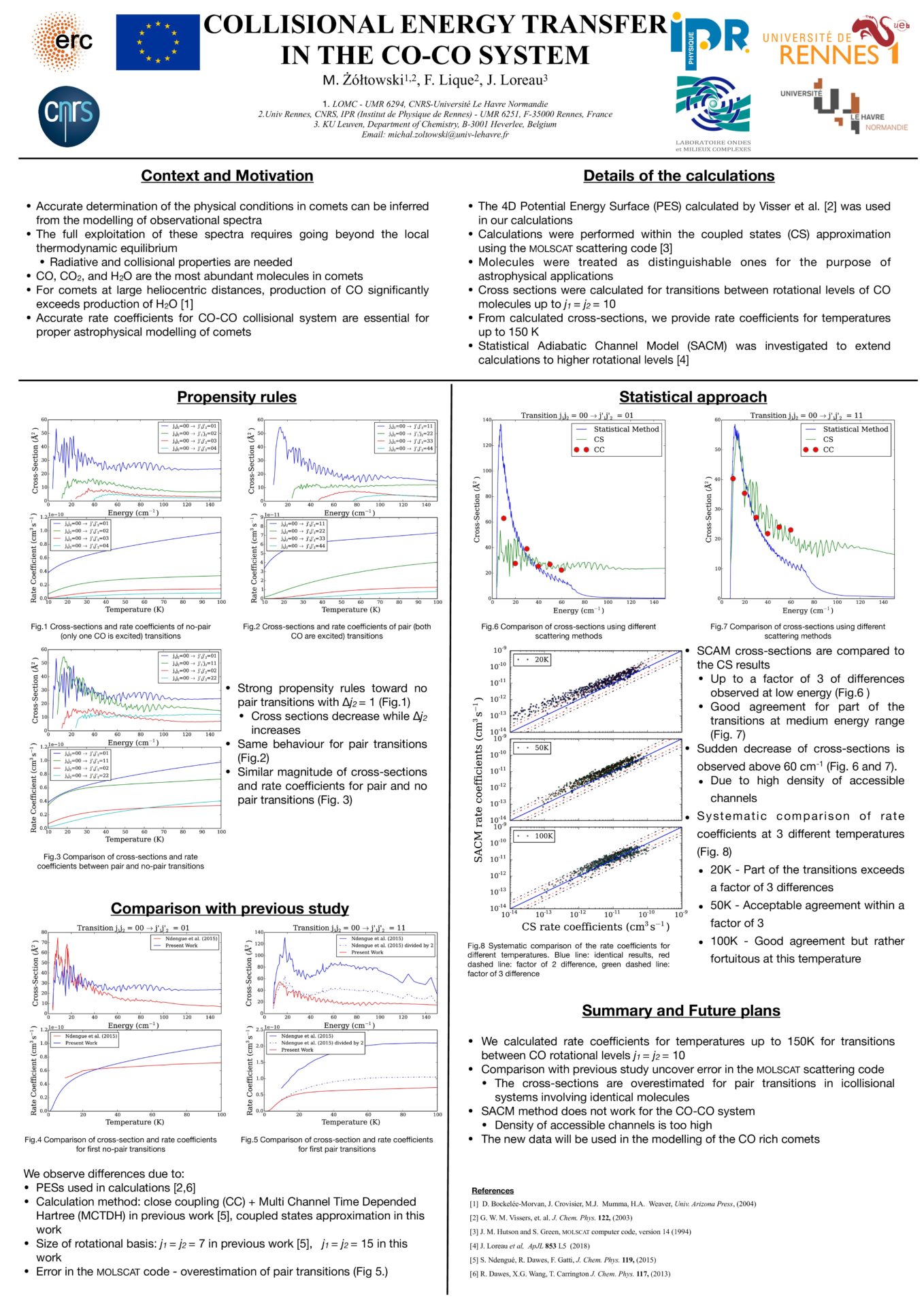
Collisional energy transfer in the CO-CO system - M. Żółtowski, F. Lique and J. Loreau
Accurate determination of the physical conditions in comets can be inferred from the modeling of molecular spectra. However, the full exploitation of molecular spectra requires to go beyond the local thermodynamic equilibrium (LTE) approach and hence requires radiative and collisions properties of the molecular species.
Among the cometary molecules, CO is of key importance, and is even the dominant species in cometary at large heliocentric distances. Here, we present new scattering calculations for the rotational excitation in CO-CO collisional system using non-reactive quantum scattering code MOLSCAT.
Calculations were performed using coupled-channel methods within the coupled states approximation, using the four dimensional potential energy surface calculated of Vissers et al. Collisional rate coefficients are provided for rotational levels up to j ≤ 10 and for temperatures up to 150 K.
The new results are compared to the previous ones from literature and significant differences are found, especially for the dominant transitions. The impact of these new data of the astrophysical modeling is also discussed.
The dynamics of the H3O+ + H2 interaction: state-to-state collisional data with astrophysical applications - S. Demes, F. Lique, A. Faure, F. van dr Talk, C. Rist and P. Hily-Blant
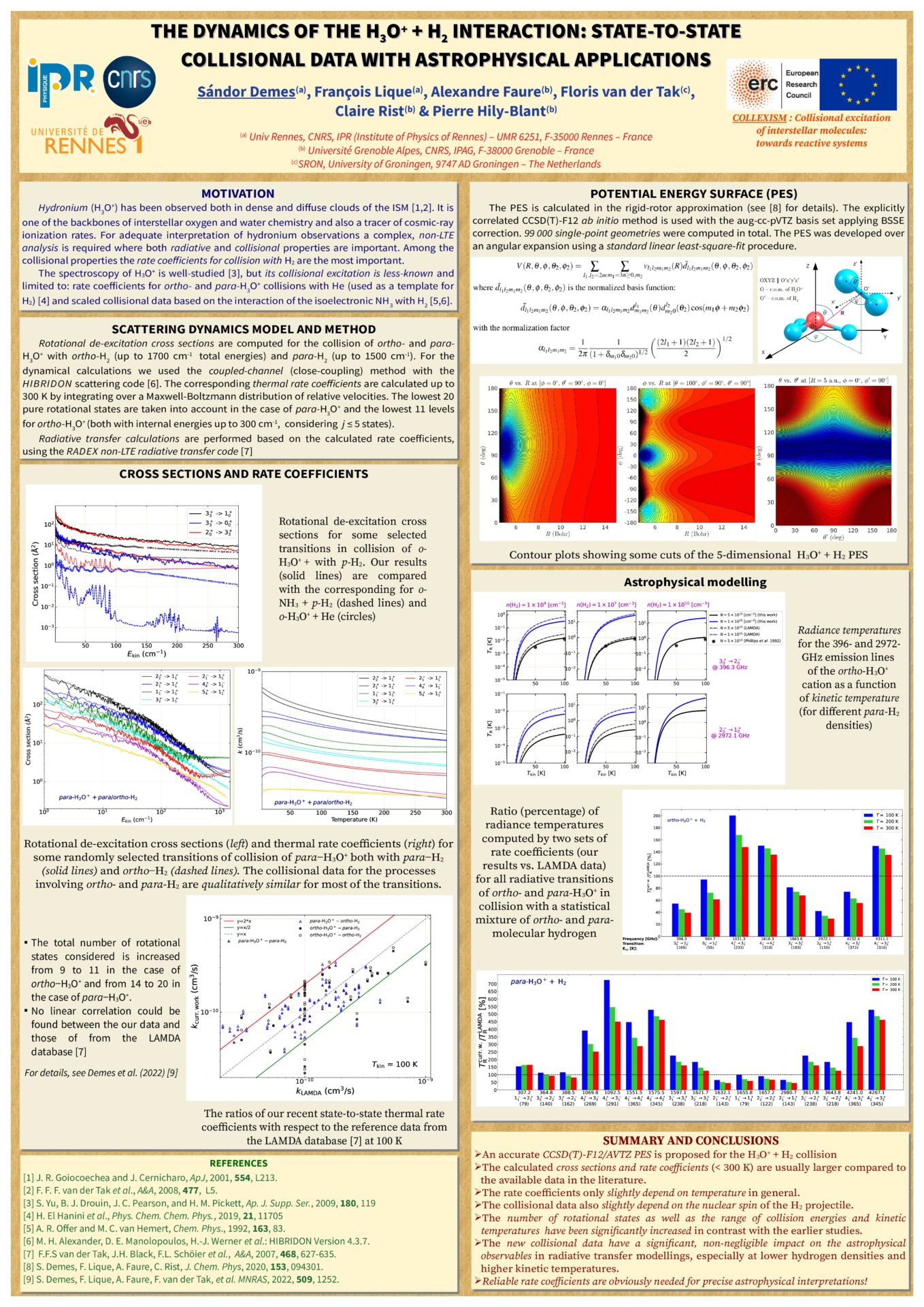
Recent astronomical observations have been showed that molecular clouds in the interstellar medium (ISM) exhibit a very rich and complex chemistry [1]. However, in order to interpret these observations, complementary research is required. So for example, to correctly describe the physico- chemical conditions in interstellar environments, where local thermodynamic equilibrium conditions are usually not fulfilled, a complex theoretical analysis is required, which involves the computation of state-to-state collisional rate coefficients for the rotational transitions of molecular species.
Hydronium (H3O+) has been detected in both dense and diffuse molecular clouds, and plays a crucial role in oxygen and water chemistry [2]. Since there are only limited works devoted to study its collisional excitation by interstellar colliders [3], accurate rate coefficients are obviously needed.
The rotational excitation of the H3O+ cation in collision with H2 molecule (the most dominant collider in the ISM) is studied for the first time [4]. State-to-state rotational de-excitation cross sections were computed using the close-coupling method, based on a highly accurate 5D potential energy surface [5]. The thermal rate coefficients were derived up to 300 K kinetic temperatures by integrating the cross sections over a Maxwell–Boltzmann distribution of velocities. The calculated rate coefficients are used then in radiative transfer modeling of hydronium excitation in interstellar molecular clouds, giving new insights into H3O+ observations and more details about water chemistry in Space.
[1] Gas-Phase Chemistry in Space, ed. F. Lique and A. Faure (IOP Publishing, Bristol, UK, 2019).
[2] F. F. S. Van Der Tak, S. Aalto, R. Meijerink, Astron. Astrophys., 477 L5 (2008).
[3] A. R. Offer and M. C. van Hemert, Chem. Phys., 163 83 (1992).
[4] S. Demes, F. Lique, F.F.S. van der Tak et al., Mon. Notices Royal Astron. Soc., 509 1252 (2022).
[5] S. Demes, F. Lique, A. Faure, C. Rist, J. Chem. Phys., 153, 094301 (2020).
Theoretical rate coefficients for the reaction between C3N with H2 and CH4 - R. Jara Toro, M. Fournier, J.-C. Guillemin, I. R. Sims and F. Lique
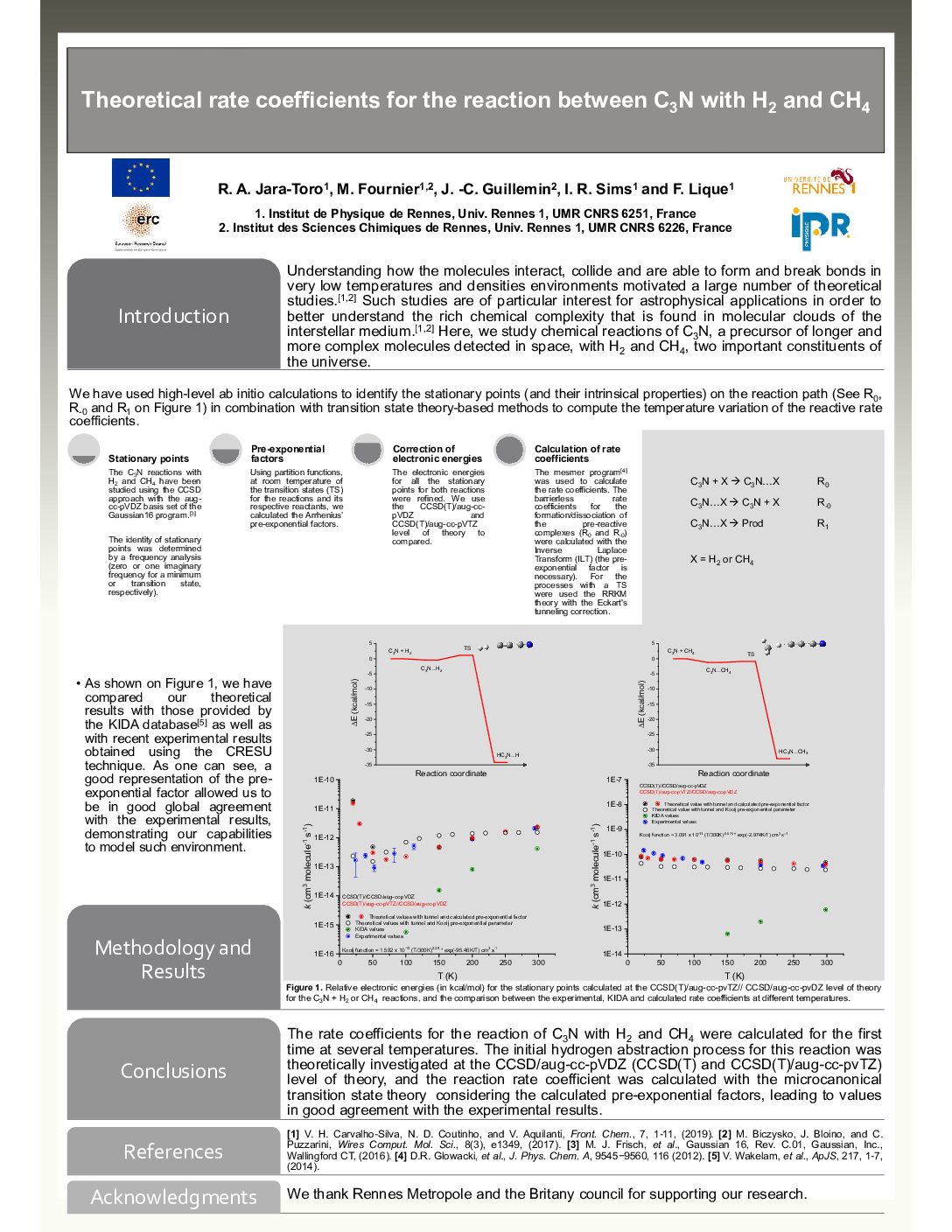
Understanding how the molecules interact, collide and are able to form and break bonds in very low temperatures and densities environments motivated a large number of theoretical studies.[1,2] Such studies are of particular interest for astrophysical applications in order to better understand the rich chemical complexity that is found in molecular clouds of the interstellar medium.[1,2] Here, we study chemical reactions of C3N, a precursor of longer and more complex molecules detected in space, with H2 and CH4, two important constituents of the universe.
We have used high-level ab initio calculations to identified the stationary points (and its intrinsical properties) on the possible reaction path (See R0, R-0 and R1 on Figure 1) in combination with transition state theory-based methods to compute the temperature variation of the reactive rate coefficients.
As shown on Figure 1, using as example the reaction between CAs shown on Figure 1, using as example the reaction between C3N with H2, we have compared our theoretical results with those provided by the KIDA database [3] as well as with recent experimental results obtained using the CRESU technique. As one can see, a good representation of the pre-exponential factor (obtained from the partition functions of the transition state and reactants) and the use of the Inverse Laplace Transform (ILT) to calculate the barrierless rate coefficients for the formation/dissociation of the pre-reactive complex (R0 and R-0) allowed us to be in good global agreement with the experimental results, demonstrating our capabilities to model such environment.
[1] V. H. Carvalho-Silva, N. D. Coutinho, and V. Aquilanti, Front. Chem., 7, 1-11, (2019).
[2] M. Biczysko, J. Bloino, and C. Puzzarini, Wires Comput. Mol. Sci., 8(3), e1349, (2017).
[3] V. Wakelam, et al., ApJS, 217, 1-7, (2014).